20251106_calderon_rotation.Rproj
apa.csl
assets
barcodes
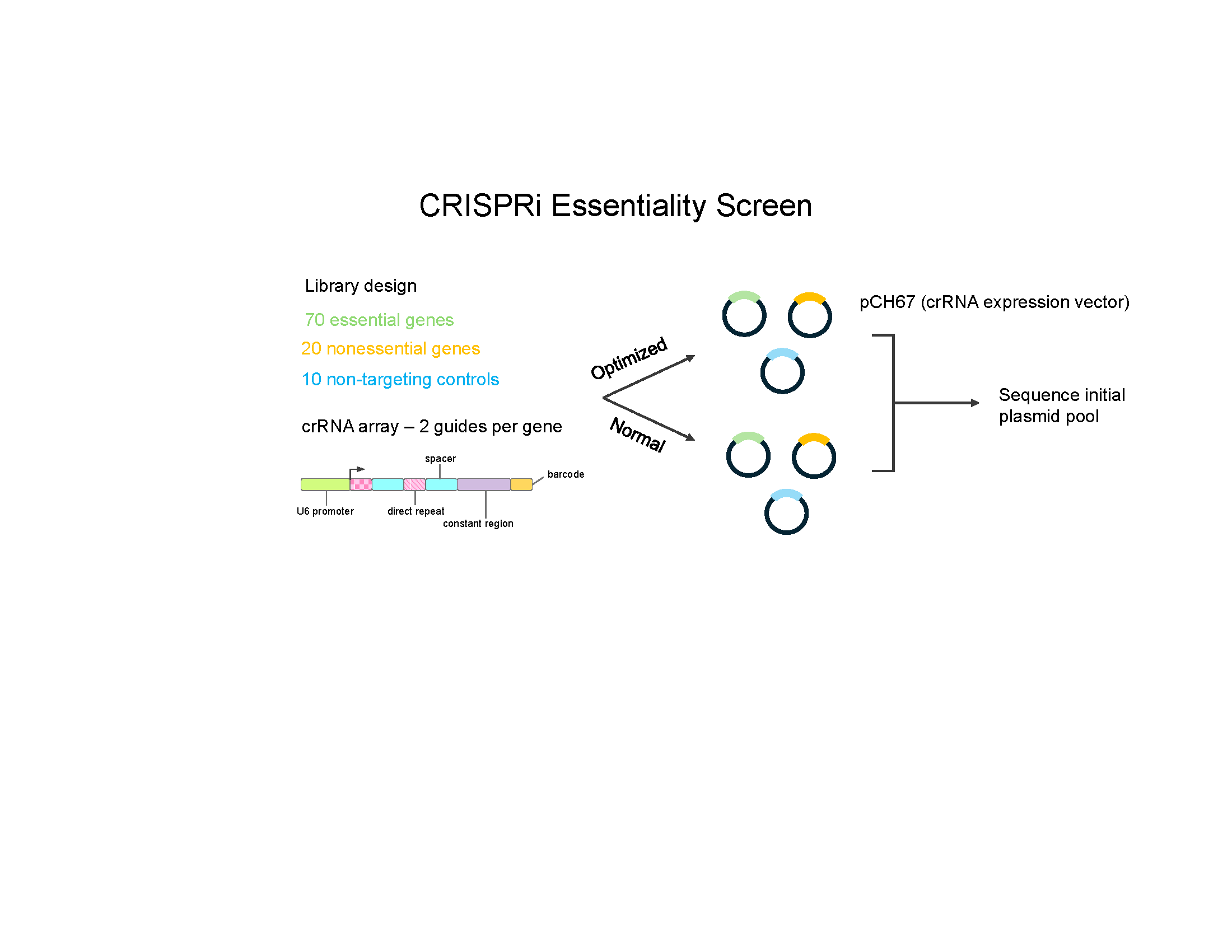
berkeley_ucsf_bioe_logo.pngLibrary design for multiplexed CRISPR screens to assess barcode swapping
Calderon Lab Meeting
November 6, 2025

Noah Gordon
PhD Student, Bioengineering
University of California, Berkeley
University of California, San Francisco
Library design for multiplexed CRISPR screens to assess barcode swapping
Calderon Lab Meeting
November 6, 2025
Noah Gordon
PhD Student, Bioengineering
University of California, Berkeley
University of California, San Francisco

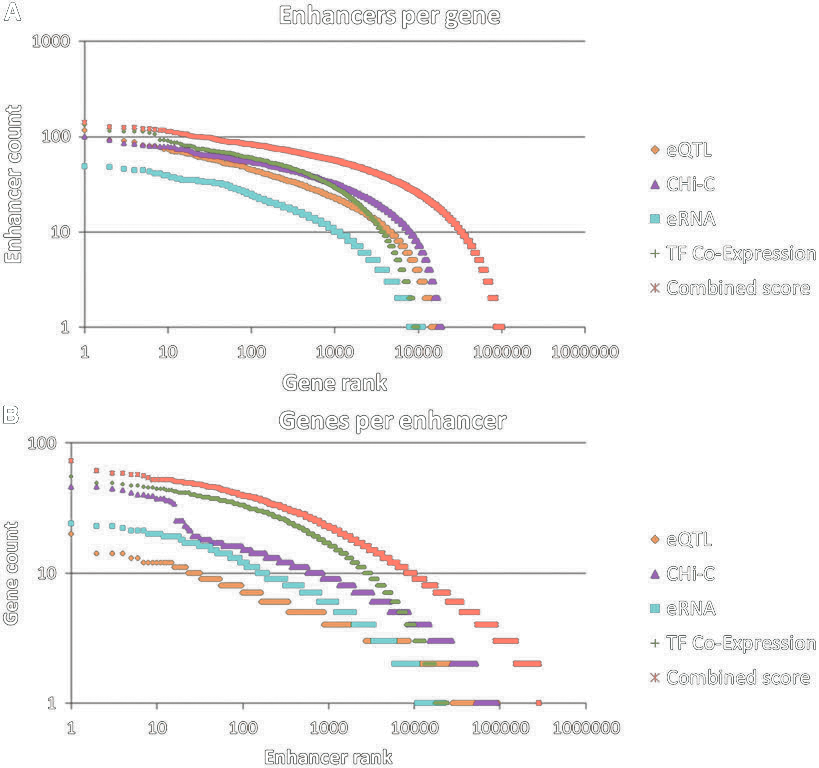

Gene-enhancer associations (Fishilevich et al., 2017)
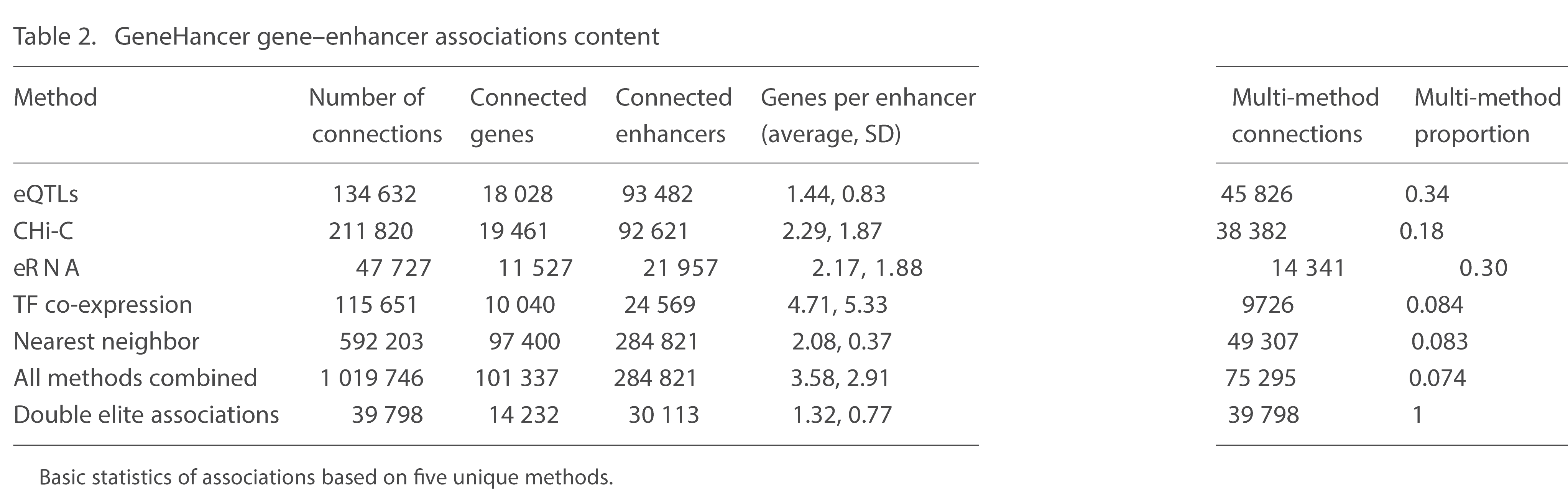
Gene-enhancer associations (Fishilevich et al., 2017)
Liebler (2014)
Cohen (2018)
“Although multiplex gene editing is possible with Cas9 nuclease, it requires relatively large constructs or simultaneous delivery of multiple plasmids, both of which are problematic for multiplex screens or in vivo applications.” (Zetsche et al., 2017)
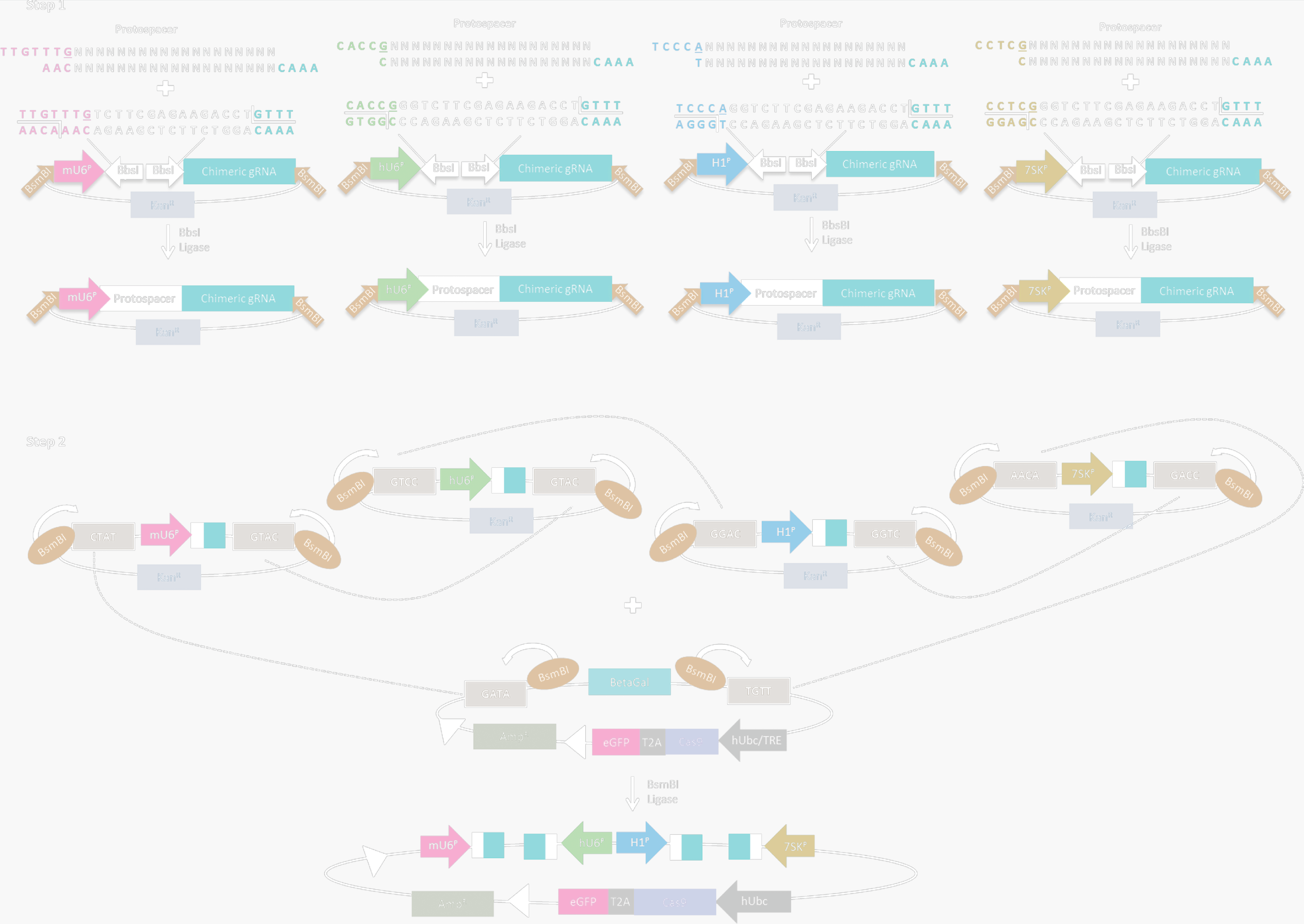
Golden Gate assembly of single lentiviral vectors encoding CRISPR/Cas9 and multiple sgRNA expression cassettes. (Kabadi, Ousterout, Hilton, & Gersbach, 2014)
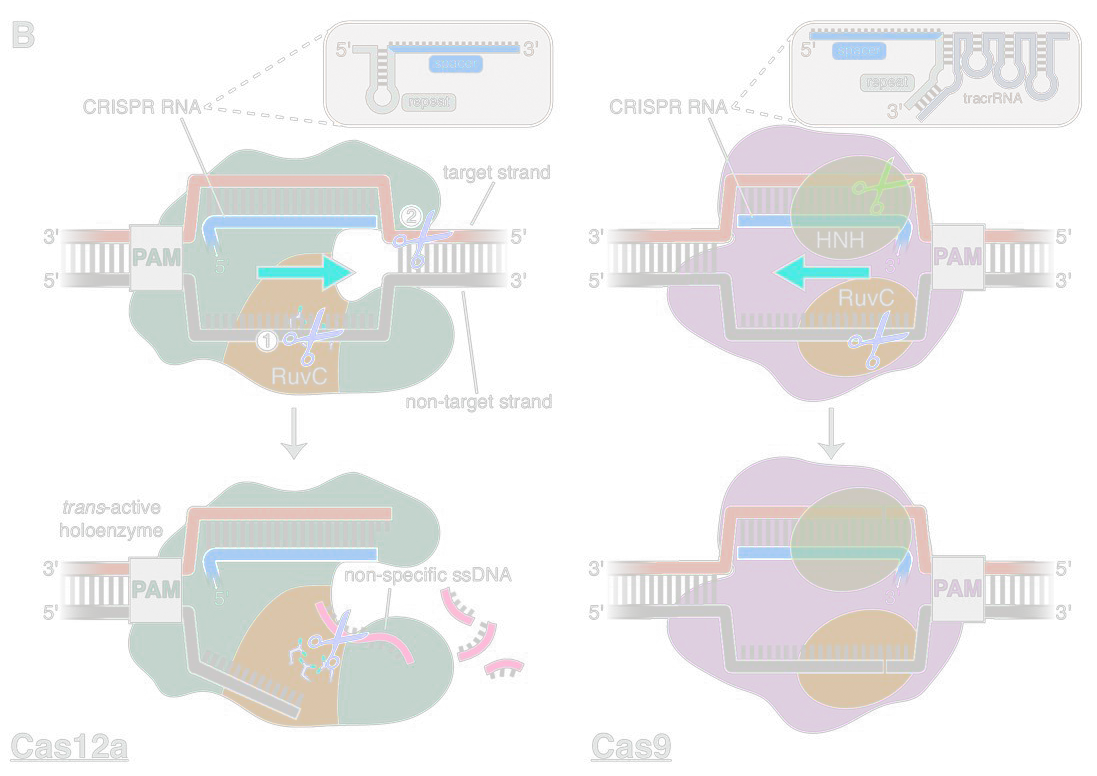
Enter Cas12A (Cofsky et al., 2020)
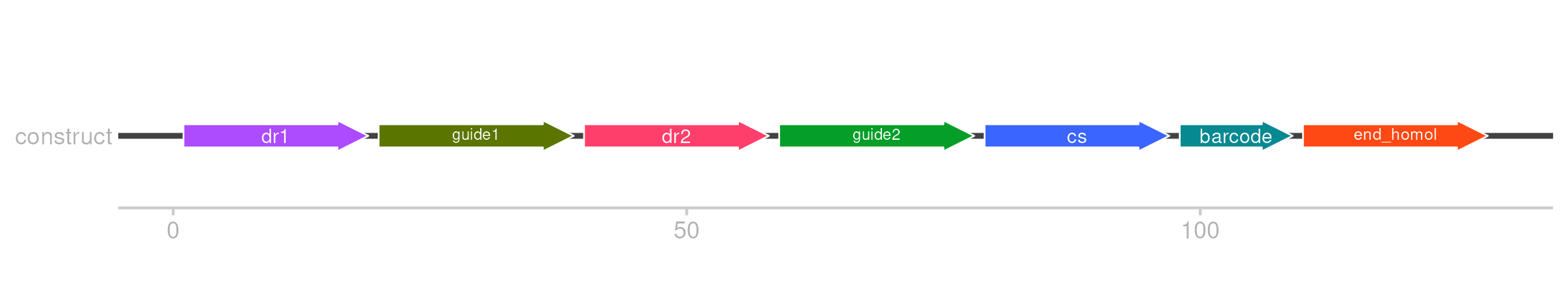
Cas12A Library
upload.sh
top_right.png
top_right_logo.png
theme
teal_stripe_background.png gene gene_effect z_score mean stddev
1 ISCU -3.299316 -3.034026 -2.017645 0.4224325depmap_K562_CRISPR_z_scores |>
filter(abs(z_score) > 3) |>
arrange(gene_effect) |>
head(71) |>
select(gene) |>
write.table("depmap_essential_genes_70.tsv",sep = "\t", row.names = FALSE)
depmap_K562_CRISPR_z_scores |>
arrange(desc(gene_effect)) |>
head(20) |>
select(gene) |>
write.table("depmap_proliferative_genes_20.tsv",sep = "\t", row.names = FALSE)
depmap_K562_CRISPR_z_scores |>
arrange(abs(gene_effect)) |>
head(30) |>
select(gene) |>
write.table("depmap_non-essential_genes_20.tsv",sep = "\t", row.names = FALSE)# CONSTANTS FROM HSUING ET AL
WTDR <- "AATTTCTACTCTTGTAGAT"
DR1 <- "AATTTCTACTGTCGTAGAT"
DR3 <- "AATTTCTACTCTAGTAGAT"
DR8 <- "AATTTCTCCTCTAGGAGAT"
DR10 <- "AATTCCTACTCTCGTAGGT"
# LIBRARY CONSTANT REGIONS
CLONING_SITE <- "GTAGATCGAGACGGTTGTATTTGTACGTCTCAAATTTCTCCTCT"
END_HOMOLOGY <- "ACTGCTTTTTGCTTGTACT"
# Database sources for barcodes, guides
#barcodes generated using https://github.com/finkelsteinlab/freebarcodes
barcodes <- readLines("barcodes/barcodes12-2.txt")
# dolcetto from https://www.addgene.org/pooled-library/broadgpp-human-crispri-dolcetto/
# dolcetto file: https://media.addgene.org/cms/filer_public/1c/59/1c59fe51-ef6e-44bf-963a-598edadcd66f/broadgpp-dolcetto-targets-seta.txt
dolcetto <- read.delim("broadgpp-dolcetto-targets-seta.txt", stringsAsFactors = FALSE)
dolcetto_guides_from_genes <- function(dolcetto_df, genes, group_name) {
dolcetto_df %>%
filter(Annotated.Gene.Symbol %in% genes$gene) %>%
select(-Annotated.Gene.ID) %>%
rename(
gene = Annotated.Gene.Symbol,
guide = Barcode.Sequence
) %>%
mutate(group = group_name) %>%
arrange(gene) %>%
group_by(gene) %>%
mutate(guide_num = row_number()) %>%
ungroup() %>%
pivot_wider(
id_cols = c(gene, group),
names_from = guide_num,
values_from = guide,
names_prefix = "guide"
) %>%
select(group, gene, starts_with("guide"))
}
depmap_70_essential <- read.table("depmap_essential_genes_70.tsv", sep = "\t", header = TRUE)
essential <- dolcetto_guides_from_genes(dolcetto, depmap_70_essential, "essential")
depmap_20_nonessential <- read.table("depmap_non-essential_genes_20.tsv", sep = "\t", header = TRUE)
nonessential <- dolcetto_guides_from_genes(dolcetto, depmap_20_nonessential, "non-essential")
ntc <- dolcetto %>%
# Filter only "NO-TARGET" rows
filter(Annotated.Gene.Symbol == "NO-TARGET") %>%
# Drop unnecessary column
select(-Annotated.Gene.ID) %>%
# Rename columns
rename(
gene = Annotated.Gene.Symbol,
guide = Barcode.Sequence
) %>%
# Add a group column (optional)
# Arrange by gene (though all are "NO-TARGET")
arrange(gene) %>%
# Assign group numbers for every 3 rows
mutate(group_id = (row_number() - 1) %/% 3 + 1) %>%
# Number guides within each group
group_by(group_id) %>%
mutate(guide_number = row_number()) %>%
ungroup() %>%
# Pivot wider so each group becomes one row
pivot_wider(
id_cols = group_id,
names_from = guide_number,
values_from = guide,
names_prefix = "guide"
) %>%
#ok now we have essential, nonessential, and ntc as dfs with these 5 columns:
#group, gene, guide1, guide2, guide3
full_library <- bind_rows(essential, nonessential, ntc)
full_library <- bind_cols( full_library, barcode = barcodes[1:nrow(full_library)])
full_library <- full_library %>%
mutate(dr1 = WTDR) %>%
mutate(dr2 = DR1) %>%
mutate(cs = CLONING_SITE) %>%
mutate(end_homol = END_HOMOLOGY ) %>%
select(group, gene, dr1, guide1, dr2, guide2, cs, barcode, end_homol)
idt_library <- full_library %>%
mutate(
sequence = paste0(dr1, guide1, dr2, guide2, cs, barcode, end_homol),
`Pool name` = "cas12_essentiality_screen" # replace with the desired pool value
) %>%
select(`Pool name`, sequence)
write_xlsx(idt_library, "idt_library_cas12_screen.xlsx")# A tibble: 6 × 7
dr1 guide1 dr2 guide2 cs barcode end_homol
<chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 AATTTCTACTCTTGTAGAT CGAGGACGGGCCGGGCACGA AATTT… CGGGC… AATT… AACAAC… AATTCCTA…
2 AATTTCTACTCTTGTAGAT GCCTTCGAGTCCTGGGGCGG AATTT… GCGGC… AATT… AACAAC… AATTCCTA…
3 AATTTCTACTCTTGTAGAT GACGCGGCGCTGCTCCATGG AATTT… GCGCG… AATT… AACAAG… AATTCCTA…
4 AATTTCTACTCTTGTAGAT CAGCCCTCGCGTGTACCTGA AATTT… CCTGA… AATT… AACACC… AATTCCTA…
5 AATTTCTACTCTTGTAGAT ACCGAAAGGGCCATGACGCG AATTT… GAAAG… AATT… AACAGG… AATTCCTA…
6 AATTTCTACTCTTGTAGAT CACCGAATTGAAGAGCATCA AATTT… GCGAT… AATT… AACCAC… AATTCCTA…# A tibble: 6 × 7
dr1 guide1 dr2 guide2 barcode cs end_homol
<chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 AATTTCTACTCTTGTAGAT CGAGGACGGGCCGGGCACGA AATTT… CGGGC… AACAAC… AATT… AATTCCTA…
2 AATTTCTACTCTTGTAGAT GCCTTCGAGTCCTGGGGCGG AATTT… GCGGC… AACAAC… AATT… AATTCCTA…
3 AATTTCTACTCTTGTAGAT GACGCGGCGCTGCTCCATGG AATTT… GCGCG… AACAAG… AATT… AATTCCTA…
4 AATTTCTACTCTTGTAGAT CAGCCCTCGCGTGTACCTGA AATTT… CCTGA… AACACC… AATT… AATTCCTA…
5 AATTTCTACTCTTGTAGAT ACCGAAAGGGCCATGACGCG AATTT… GAAAG… AACAGG… AATT… AATTCCTA…
6 AATTTCTACTCTTGTAGAT CACCGAATTGAAGAGCATCA AATTT… GCGAT… AACCAC… AATT… AATTCCTA…
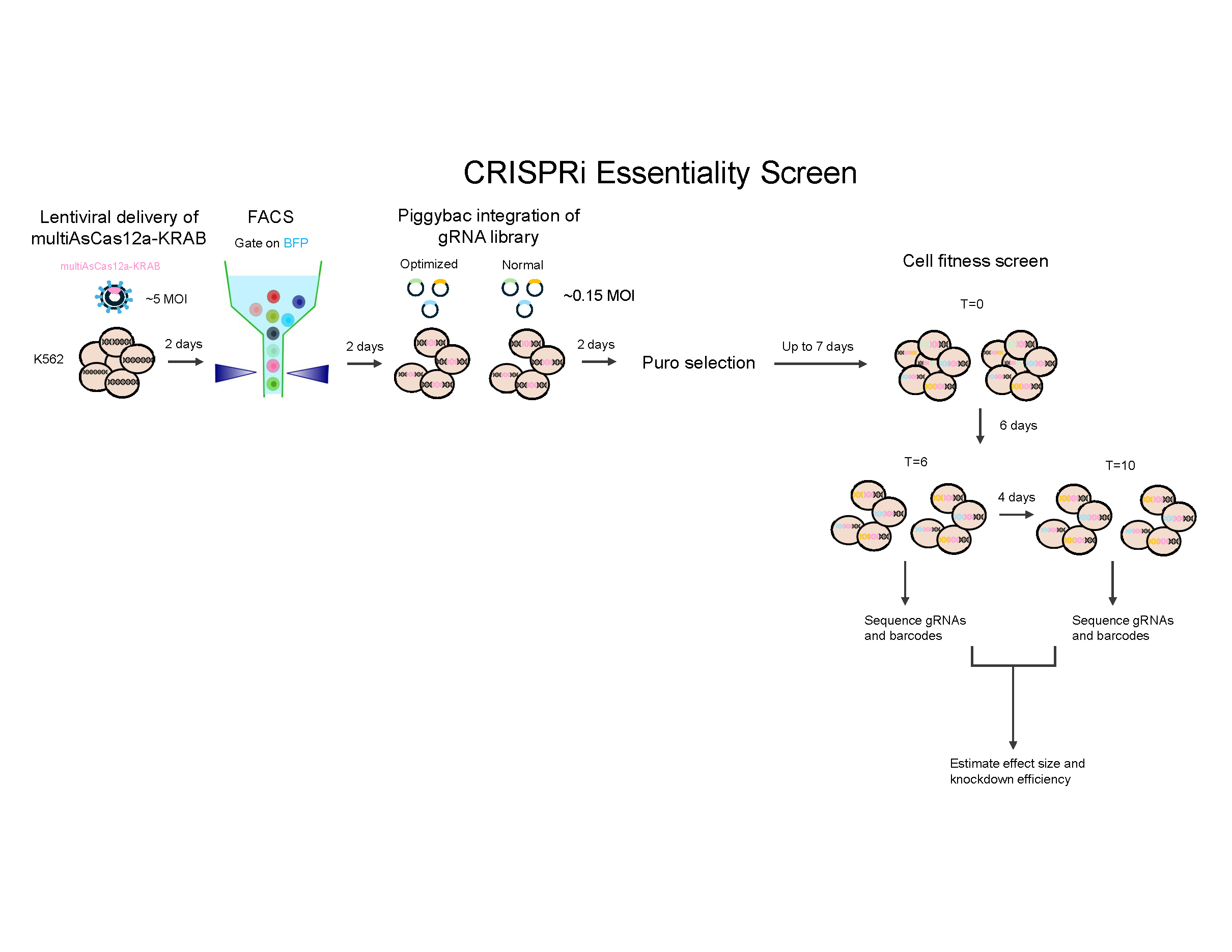
Cas12 Pilot
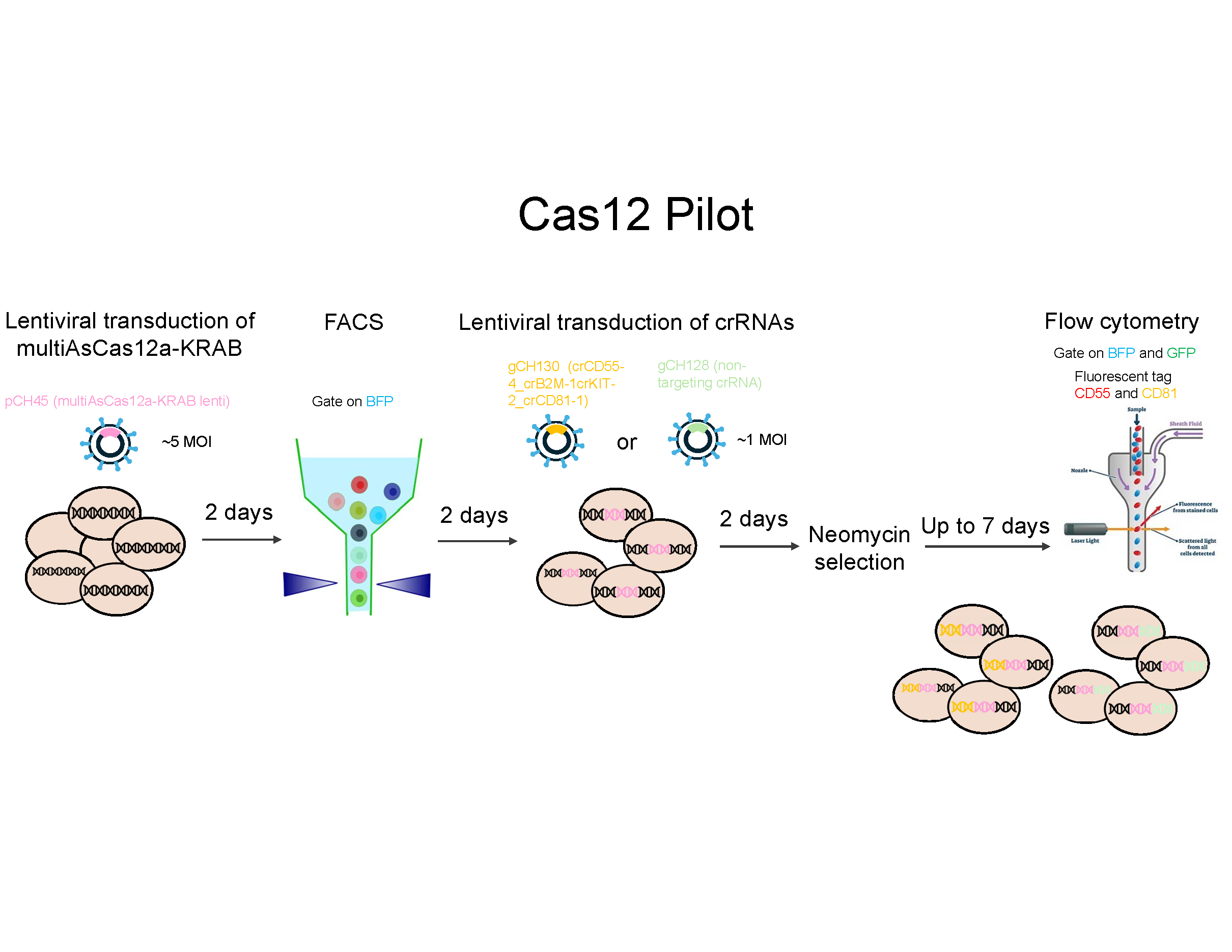
Show flow data
What’s next?
Thank you!


Source: Diego
[1]. Cofsky, J. C., Karandur, D., Huang, C. J., Witte, I. P., Kuriyan, J., & Doudna, J. A. (2020). CRISPR-Cas12a exploits r-loop asymmetry to form double-strand breaks. Elife, 9, e55143.
[2]. Cohen, J. (2018). CRISPR too fat for many therapies — so scientists are putting the genome-editor on a diet. Science. Retrieved from https://www.science.org/content/article/crispr-too-fat-many-therapies-so-scientists-are-putting-genome-editor-diet
[3]. Farh, K. K.-H., Marson, A., Zhu, J., Kleinewietfeld, M., Housley, W. J., Beik, S., Shoresh, N., Whitton, H., Ryan, R. J. H., Shishkin, A. A., Hatan, M., Carrasco-Alfonso, M. J., Mayer, D., Luckey, C. J., Patsopoulos, N. A., De Jager, P. L., Kuchroo, V. K., Epstein, C. B., Daly, M. J., Hafler, D. A., & Bernstein, B. E. (2015). Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature, 518(7539), 337–343. https://doi.org/10.1038/nature13835
[4]. Fishilevich, S., Nudel, R., Rappaport, N., Hadar, R., Plaschkes, I., Iny Stein, T., Rosen, N., Kohn, A., Twik, M., Safran, M., & others. (2017). GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database, 2017, bax028.
[5]. Kabadi, A. M., Ousterout, D. G., Hilton, I. B., & Gersbach, C. A. (2014). Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Research, 42(19), e147–e147.
[6]. Liebler, J. (2014). CRISPR–Cas9 in complex with guide RNA and target DNA. Online image, Art of the Cell. Retrieved from https://www.artofthecell.com/wp-content/uploads/2014/06/Art-of-the-Cell-CRISPR-Cas9-in-Complex-with-Guide-RNA-and-target-DNA.jpg?w=1920&ssl=1
{kind=link}
[7]. Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., Winblad, N., Choudhury, S. R., Abudayyeh, O. O., Gootenberg, J. S., & others. (2017). Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nature Biotechnology, 35(1), 31–34.